






聚良材睿智,为迈向一流而奋斗

近日,浙江大学海南研究院樊龙江教授团队在Briefings in Bioinformatics上发表了题为 mKmer: An unbiased K-mer embedding of microbiomic single-microbe RNA sequencing data的研究论文。该研究开发了一种基于K-mer的无偏微生物组单细胞或单菌RNA(smRNA)表达矩阵构建新算法,在无需参考基因组情况下,可以对人类肠道、土壤等复杂微生物组样本smRNA数据进行准确的降维聚类分析,利用K-motif对微生物分类特异性、潜在功能等进行预测。团队开发了相应的生物信息学工具mKmer(https://github.com/bioinplant/mKmer/),供研究人员方便使用。

微生物单细胞转录组测序技术(smRNA-seq)的飞速发展为揭示了微生物分类和状态异质性提供了强有力工具,对生物学研究带来深远影响。同时,smRNA-seq大数据分析面临着巨大挑战:如何构建高质量的基因表达矩阵以支持下游分析?由于环境微生物或微生物组样本物种构成的复杂性,目前还无法获得一个完整和精确的参考基因组,特别是微生物组样本中许多物种缺乏参考基因组或无高质量基因注释。为此,樊龙江教授团队开发了不依赖参考基因组的分析新方法mKmer,在人类肠道和土壤等环境样本中取得了很好效果(图1),由此解决了上述分析难题。
mKmer提供了七个算法模块,包括确定最优K值、识别关键保守序列等。分析框架中,通过筛选拐点前的高频保守K-mer(HCK)、构建表达矩阵并进行物种鉴定与功能分析。HCK解决了因K-mer种类过多导致的计算时间与高维矩阵问题,显著提升了mKmer的性能与实用性。由于无需任何参考基因组,测序读段的利用率达到了100%;同时,通过将HCK转化为氨基酸序列(基序)进行功能注释。
与基于参考基因集的基因表达矩阵比较,基于K-mer的聚类区分度与准确性均显著提升(图1)。目前传统方法会大幅削减微生物多样性及丰度,低估真实生态复杂性。我们采用戴维斯-鲍尔丁指数(DBI)与轮廓系数(SC)定量评估七组测试smRNA数据,结果显示mKmer的DBI更低,SC更高,聚类效果更优。
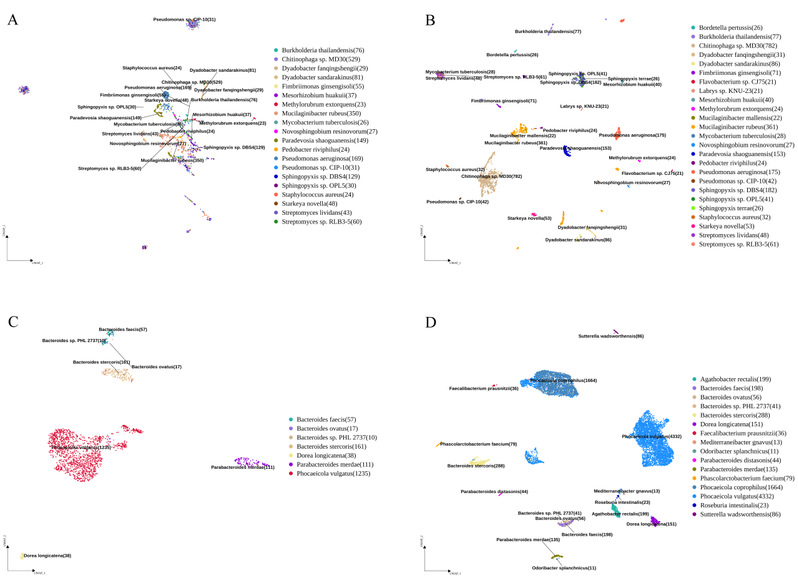
浙江大学海南研究院博士生莫钫宇为论文的第一作者,浙江大学樊龙江教授、王永成研究员等为通讯作者。三亚华大生命科学研究院白寅琪、浙江大学附属第一医院沈一飞等参与相关研究。该研究得到了海南省教育厅博士科研创新基金资助。
论文链接:https://doi.org/10.1093/bib/bbaf227
|
Copyright 2003-2020 All Rights Reserved版权所有:浙江大学海南研究院 浙ICP备05074421号-1 |
